ILLUMINA LIBRARIES
Please acknowledge the Diabetes Genomics Analysis Core in all publications resulting from full or partial use of our services, as this supports future Diabetes Research Center grant renewals.
“Data was obtained through the Diabetes Genomics Analysis Core facility of the Stanford Diabetes Research Center, supported by the NIH/NIDDK under Award Number NIH P30 DK116074.”
NGS Library Preparation
Total RNA
Total RNA-Seq provides transcriptional profiling for both the coding and noncoding regions of the genome. Ribo-zero depletion of rRNA is followed by strand-specific library preparation using the TruSeq Stranded Total RNA Library Preparation Kit.
Sample Requirements: Minimum of 500 ng purified RNA at a concentration of 50 ng/ul. A260/280 > 2. RIN < 8 is acceptable.
mRNA
mRNA-Seq provides transcriptional profiling for the coding portion of the genome. Purification of poly-A containing mRNA molecules using oligo-DT attached magnetic beads is followed by strand-specific library preparation using the TruSeq Stranded mRNA Library Preparation kit.
Sample Requirements: Minimum of 500 ng purified RNA at a concentration of 10 ng/ul. A260/280 > 2. RIN > 8.
Fluidigm C1 Single-Cell cDNA Libraries
Single-Cell RNA-Seq Following single-cell capture and conversion of mRNA into cDNA on the Fluidigm C1 Single-Cell AutoPrep System, RNA sequencing libraries are generated using a modified Illumina Nextera XT DNA sample preparation protocol.
Sample Requirements: cDNA normalized to 0.2 ng/ul in a 96-well plate with volume exceeding 3 ul.
Chromium Genome Solution
The Chromium Genome uses GemCode technology to partition a high molecular weight genomic DNA sample across up to millions of GEMs. As a result of partitioning, linked reads are generated containing a unique 10x Barcode that maps back to the original HMW gDNA that it originated from. This long-range information has been adopted in many applications including whole-genome phasing, structural variant analysis, and de novo genome assembly.
Sample Requirements: Minimum of 100 ng of HMW gDNA (>50 kb), A260/280 > 1.8. Profile from TapeStation genomic DNA or similar assay must be provided with submission.
Chromium Single Cell 3' Solution
The Chromium Single Cell 3’ leverages 10x’s GemCode technology to provide transcriptional profiling of 1,000s to 10,000s of individual cells. Transcriptional profiling at the single cell level rather than a bulk sample allows for deconvolution of heterogenous samples.
Sample Requirements: Inquire for customized requirements.
Whole-Genome Bisulfite Sequencing
Whole-Genome Bisulfite Sequencing can be used to detect patterns of cytocine methylation in the genome. Bisulfite conversion of DNA is completed using the Zymo Research EZ DNA Methylation-Gold Kit followed by library preparation using the EpiGnome/TruSeq DNA Methylation kit.
Sample Requirements: Minimum of 200 ng purified DNA at a concentration of 10 ng/ul. A260/280 > 1.8.
PacBio SMRTbell Libraries
SMRTbell library preparation does not utilize amplification steps; the completed library molecules are used as direct templates for the sequencing process. Due to this, the quality of the starting DNA has enormous influence on the success and quality of the sequencing results. High-quality, high-molecular-weight genomic DNA is extremely important for obtaining long reads and optimal performance.
Gel image submitted with sample showing no signs of degradation. For amplicon or cDNA samples, a Bioanalyzer trace can be submitted instead.
Nanodrop readings:
260/280: ratio should be around ~1.8; higher values are acceptable, low values indicate contamination/very low concentration.
260/230: ratio should be ~2.0-2.2
Sample Concentration:
Use Qubit or PicoGreen for best results
Submit a minimum of 1.5 ug for <2kb libraries, 2.5 ug for 5 kb libraries, 15 ug for 20kb+
Volume and Buffers:
Max volume 130 uL
Avoid buffers using EDTA for DNA resuspension
Additonal Best Practices can be found here
Illumina Sequencing
Please support the DGA Core by acknowledging our services in your publications.
If you use the HiSeq 4000, please acknowledge NIH Grant # 1S10OD020141-01
Thanks for your help!
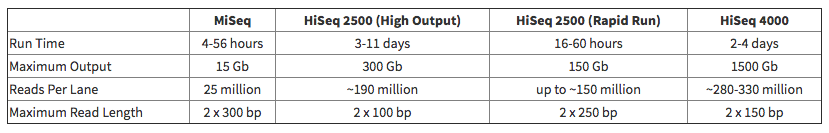
The Illumina HiSeq 4000 is over three times faster than an identical run length on the HiSeq 2000 while providing over 60% more reads per lane. However, the 4000 has more stringent library requirements and requires a larger sample size than the 2000.
A 2×101 run will take about 2-3 days to complete; a 2×151 will take 3-4. (Time variation can occur depending on the time of day clustering was performed: in the morning or afternoon).
The HiSeq 4000 generally gives 280-330 million reads per lane for a flowcell total of around 2.2 billion reads. While the patterned flow cell used on the 4000 is fixed at 480 million wells/reads per lane, Illumina recommends between 60-70% passed filter (PF) to maximize the number of unique reads in the lane. A higher PF will provide a higher amount of non-unique reads, which means more duplicate data. The HiSeq 4000 is biased towards smaller fragment sizes, so please try to keep your samples close to a consistent fragment length.
More information on the HiSeq 4000 specifications can be found on Illumina's website (PDF).
The Illumina HiSeq 2500 allows more versatility in types of runs than the HiSeq 4000. As many clients are producing libraries that haven't been optimized or cleared for use on the 4000, the 2000/2500 are good substitutes. A 2x101 run will take about 11 days to complete; a 1x50 run will take 3 days (plus additional time for clustering). The HiSeq 2500 generally gives 190 million reads per lane for a flow cell total of around 1.5 billion reads.
More information on the HiSeq 2500 specifications can be found on Illumina's website (PDF).
Rapid Run Mode
Illumina indicates that the Rapid flowcells can get up to ~150 million paired reads per lane under optimal conditions.
Rapid Run Mode is great for libraries that are not optimized for the 4000, require more data than a MiSeq provides, and need the results quicker than a 2500 high throughput mode will allow.
One of the great advantages to the MiSeq is the quick turnaround time for runs. The MiSeq is capable of long reads, making it great for de novo assembly of small genomes. The MiSeq is also great for QC tests on sequencing workflows before committing to larger batches on more expensive machines.
The MiSeq v2 kit provides 12-15 million single reads or 24-30 million paired-end reads, while the MiSeq v3 kit provides 22-25 million single reads or 44-50 million paired-end reads.
More information on the MiSeq specifications can be found on Illumina's website (PDF).
Please support the DGA Core by acknowledging our services in your publications.
If you use the HiSeq 4000, please also acknowledge NIH Grant # 1S10OD020141-01
Thanks for your help!
Pacific Biosciences Sequencing
The new Pacific Biosciences Sequel system builds upon their Single-Molecule, Real-Time (SMRT) technology and delivers higher throughput, increased scalability and lower sequencing costs compared to the PacBio RS II.
The Sequel system utilizes the DNA sample as a direct template for the sequencing reaction in order to sequence individual DNA molecules in real time. Ligation of hairpin adapters converts the double-stranded DNA molecules into a circular template that enables continuous sequencing of both the forward and reverse strands without the need for amplification. The unique circular structure allows for continuous long reads of large-insert libraries or circular consensus sequencing of smaller insert sizes. Extra-long read lengths and high consensus accuracy make the Sequel a helpful tool for finding SNPs and sequencing through GC-rich areas of the genome.
Rapid, cost-effective generation of high-quality, de novo whole genome assembly
No PCR bias, uniform coverage for targeted sequencing
Direct base modification detection for epigenetic studies
Long read lengths
Single-molecule resolution of complex populations
High consensus accuracy
The basic sequencing "unit" for the Sequel is the SMRTcell, analogous to a flowcell in Illumina sequencing. SMRTcells do not have lanes, unlike flowcells, so each sample is sequenced on its own SMRTcell. There are no "read lengths" like in Illumina sequencing; instead, you choose a length of time to continuously run the sequencing reaction, known as a "movie time". The maximum movie time for the Sequel is 10 hours.
There are two different methods of loading the SMRTcell: diffusion loading and Magbead loading. Fragment size largely determines which loading strategy you will use. For smaller fragment sizes (<7.5kb), diffusion loading is generally the recommended method. In diffusion loading, SMRTbell library molecules with bound polymerase are immobilized and diffuse to the bottom of the ZMW wells after removal of excess polymerase and primer with cleanup beads. For fragments >7.5kb, Magbead loading is recommended, which removes adapter dimers and short insert sizes to maximize read lengths.
Bioinformatics Analysis
2800+ cores and 7+ Petabytes of high performance storage
Architecture specifically suited to large scale genomics data analysis but also supports general scientific computing
500+ bioinformatics software packages installed and ready to use
Specialized data analysis solution, Galaxy, available to users. Inquire for ongoing pilot programs.
Payable on an hourly basis
We not only support analysis for popular NGS data types such as RNASeq, ChIPSeq, MethylSeq, Whole Genome/Whole Exome Seq, CancerSeq, and Microbiome, we also support new data types like Hi-C, and ATAC-Seq.
Along with Secondary analysis, we provide consulting in quality control, downstream tertiary analysis, data interpretation and visualization. Custom/novel development is available.
Cloud services backed by SU agreements
Security compliance set up by DGAC system admins
Services support SU mandated security requirements e.g. two-factor authentication
On-premise and Cloud gateway support NIH dbGaP security best-practices. Learn more about dbGaP here.
Regular Stanford IT audits to keep up with security compliance requirements